To classify proteins using the CATH and SCOP databases.
THEORETICAL BACKGROUND:
Proteins (also known as polypeptides) are organic compounds made of amino acids arranged in a linear chain and folded into a globular form. The amino acids in a polymer chain are joined together by the peptide bonds between the carboxyl and amino groups of adjacent amino acid residues.
Most proteins fold into unique 3-dimensional structures. The shape into which a protein naturally folds is known as its native conformation. To describe the complicated macromolecular structure of proteins, biochemists have, for convenience, recognized four basic structural levels of organization of proteins based on the degree of complexity of their molecule. These structural levels were first described by Linderstrom-Lang and are often referred to as primary, secondary, tertiary and quaternary. The first three structural levels can exist in molecules composed of a single polypeptide chain whereas the last one involves interaction of polypeptides within a muiti-chained protein molecule.
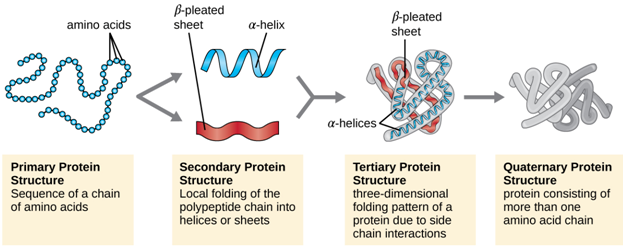
1. Primary structure :
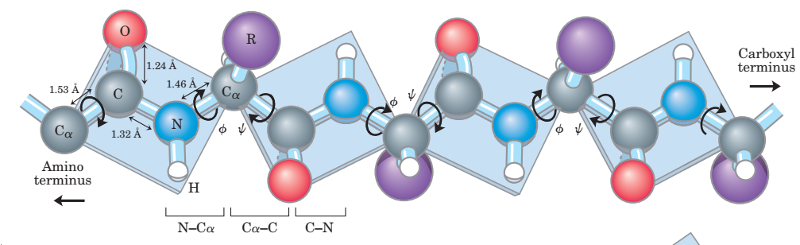
The sequence of the different amino acids constitutes the primary structure of the peptide or protein. Counting of residues always starts at the N-terminal end (NH2-group), which is the end where the amino group is not involved in a peptide bond. The primary structure of a protein encodes its uniquely folded 3D conformation. The most important factor governing the folding of a protein into 3D structure is the distribution of polar and non-polar side chains. Folding is driven by the burial of hydrophobic side chains into the interior of the molecule so as to avoid contact with the aqueous environment.
2. Secondary structure:
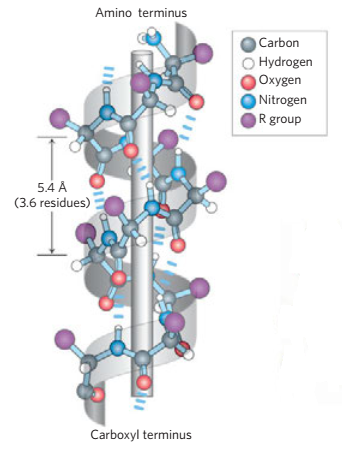
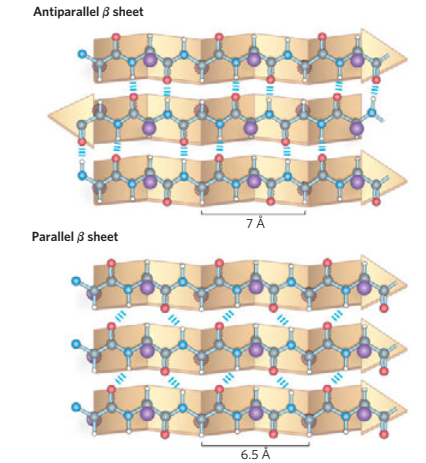
Generally, proteins have a core of hydrophobic residues surrounded by a shell of hydrophilic residues. Since the peptide bonds themselves are polar they are neutralized by hydrogen bonding with each other when in the hydrophobic environment. This gives rise to regions of the polypeptide that form regular 3D structural patterns called 'secondary structure'. There are two main types of secondary structure are the alpha helix and the beta sheet. Each of these two secondary structure elements have a regular geometry, meaning they are constrained to specific values of the dihedral angles ψ and φ . Both the alpha helix and the beta-sheet represent a way of saturating all the hydrogen bond donors and acceptors in the peptide backbone. These secondary structure elements only depend on properties of the polypeptide main chain, explaining why they occur in all proteins. The part of the protein that is not in a regular secondary structure is said to be a "non-regular structure" (not to be mixed with random coil, an unfolded polypeptide chain lacking any fixed three-dimensional structure).
Other helices, such as the 310 helix and π helix, are calculated to have energetically favorable hydrogen-bonding patterns but are rarely if ever observed in natural proteins except at the ends of α -helices due to unfavorable backbone packing in the center of the helix.
Other extended structures such as the polyproline helix and alpha sheet are rare in native state proteins but are often hypothesized as important protein folding intermediates. Tight turns and loose, flexible loops link the more "regular" secondary structure elements. The random coil is not a true secondary structure, but is the class of conformations that indicate an absence of regular secondary structure.
Amino acids vary in their ability to form the various secondary structure elements. Proline and glycine are sometimes known as "helix breakers" because they disrupt the regularity of the a-helical backbone conformation; however, both have unusual conformational abilities and are commonly found in turns.
Amino acids that prefer to adopt helical conformations in proteins include methionine, alanine, leucine, glutamic and lysine ("MALEK" in amino-acid 1- letter codes); by contrast, the large aromatic residues (tryptophan, tyrosine and phenylalanine) and C β -branched amino acids (isoleucine, valine, and threonine) prefer to adopt β -strand conformations. However, these preferences are not strong enough to produce a reliable method of predicting secondary structure from sequence alone.
Protein super secondary structures - structural motifs
Some simple combinations of secondary structure elements have been found to frequently occur in-protein structure and are referred to as 'super-secondary structure' or motifs. Structure motifs usually consist of just a few elements, e.g. the 'helix-turn-helix' has just three. However, while the spatial sequence of elements is the same in all instances of a motif, they may be encoded in any order within the underlying gene.
Protein structural motifs often include loops of variable length and unspecified structure, which in effect create the "slack" necessary to bring together in space two elements that are not encoded by immediately adjacent DNA sequences in a gene. Even when two genes encode secondary structural elements of a motif in the same order, nevertheless they may specify somewhat different sequences of amino acids. This is true not only because of the complicated relationship between tertiary and primary structure, but also because the size of the motif elements varies from one protein and the next. Examples of motifs in proteins :
Β-hairpin motif:
Consists of two adjacent antiparallel β-strands joined by a small loop. It is present in most antiparallel β structures both as an isolated ribbon and as part of more complex β-sheets.

β-α-β Motif: (a)
Frequently used to connect two parallel β-strands. The central α-helix connects the C-termini of the first strand to the N-termini of the second strand, packing its side chains against the β-sheet and therefore shielding the hydrophobic residues of the β-strands from the surface.
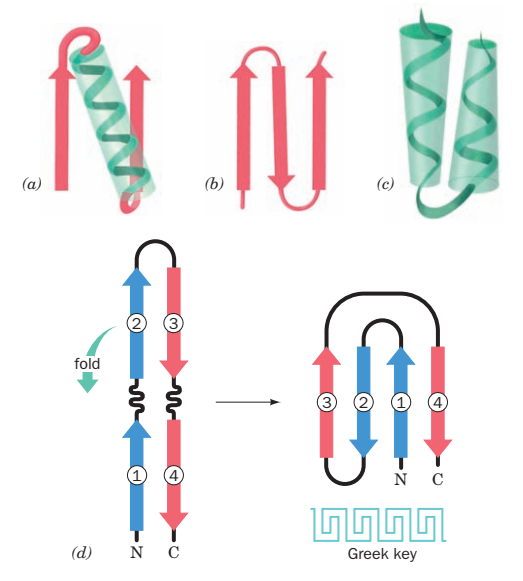
Greek Key: (d)
4 beta strands folded over into a sandwich shape.
Helix-loop-helix :(c)
Consists of alpha helices bound by a looping stretch of amino acids. Important in DNA binding proteins.
Zinc finger :(a)
Two beta strands with an alpha helix end folded over to bind a zinc ion. This motif is seen in transcription factors.
3. Tertiary structure :
The elements of secondary structure are usually folded into a compact shape using a variety of loops and turns to form the tertiary structure of protein. The formation of tertiary structure is usually driven by non-local interactions, most commonly the formation of a hydrophobic core by the burial of hydrophobic residues, but other interactions such as hydrogen bonding, ionic interactions and disulphide bonds and even post-translational modifications can also stabilize the tertiary structure. The tertiary structure encompasses all the noncovalent interactions that are not considered secondary structure, and is what defines the overall fold of the protein, and is usually indispensable for the function of the protein.
The term "tertiary structure" is often used as synonymous with the term fold. And the tertiary structure is what controls the basic function of the protein. During protein folding to tertiary level, several motifs pack together to form compact, local, semi-independent units called domains. This overall 3D structure of the polypeptide chain is referred to as the protein's 'tertiary structure'. Domains are the fundamental units of tertiary structure, each domain containing an individual hydrophobic core built from secondary structural units connected by loop regions. The packing of the polypeptide is usually much tighter in the interior than the exterior of the domain producing a solid-like core and a fluid-like surface. In fact, core residues are often conserved in a protein family, whereas the residues in loops are less conserved, unless they are involved in the protein's function. Protein tertiary structure can be divided into four main classes based on the secondary structural content of the domain :
- All- α domains have a domain core built exclusively from α -helices. This class is dominated by small folds, many of which form a simple bundle with helices running up and down.
- All- β domains have a core comprising of antiparallel (3-sheets, usually two sheets packed against each other. Various patterns can be identified in the arrangement of the strands, often giving rise to the identification of recurring motifs, for example the Greek key motif.
- α+β domains are a mixture of all-α and all-β motifs. Classification of proteins into this class is difficult because of overlaps to the other three classes and therefore is not used in the CATH domain database.
- α/β domains are made from a combination of β-α-β motifs that predominantly form a parallel β-sheet surrounded by amphipathic α-helices. The secondary structures are arranged in layers or barrels.
The CATH domain database classifies domains into approximately 800 fold families, ten of these folds are highly populated and are referred to as 'superfolds'. Super-folds are defined as folds for which there are at least three structures without significant sequence similarity. The most populated is the α / β -barrel super-fold described previously.
Domains have limits on size. The size of individual structural domains varies from 36 residues in E-selectin to 692 residues in lipoxygenase-1, but the majority, 90%, have less than 200 residues with an average of approximately 100 residues. Very short domains, less than 40 residues, are often stabilized by metal ions or disulphide bonds. Larger domains, greater than 300 residues, are likely to consist of multiple hydrophobic cores. Domains are the common material used by nature to generate new sequences, they can be thought of as genetically mobile units, referred to as 'modules'.
4. Quaternary structure :
Quaternary structure is the structure formed by several protein molecules (polypeptide chains), usually called protein subunits, which function as a single protein complex. The individual subunits are usually not covalently connected, but might be connected by a disulphide bond. Not all proteins have quaternary structure, since they might be functional as monomers. The quaternary structure is stabilized by the same range of interactions as the tertiary structure. Complexes of two or more polypeptides (i.e. multiple subunits) are called multimers. Specifically, it would be cafled a dimer if it contains two subunits, a trimer if it contains three subunits, and a tetramer if it contains four subunits. The subunits are usually related to one another by symmetry axes, such as a 2-fold axis in a dimer. Multimers made up of identical subunits may be referred to with a prefix of "homo-" (e.g. a homotetramer) and those made up of different subunits may be referred to with a prefix of "hetero-" (e.g. a heterotetramer, such as the two alpha and two beta chains of haemoglobin). Changes in quaternary structure can occur through conformational changes within individual subunits or through reorientation of the subunits relative to each other. It is through such changes, which underlie cooperativity and allostery in "multimeric" enzymes, that many proteins undergo regulation and perform their physiological function.
Many proteins share structural similarities, reflecting, in some cases, common evolutionary origins. The evolutionary-process involves substitutions, insertions and deletions in amino acid sequences. For distantly related proteins, such changes can be extensive, yielding folds in which the numbers and orientations of secondary structures vary considerably. However, where, for example, the functions of proteins are conserved, the structural environments of critical active site residues are also conserved.
In an attempt to better understand sequence/structure relationships and the underlying evolutionary processes that give rise to different fold families, a variety of structure classification schemes have been established. The nature of the information presented by a structure classification scheme is entirely dependent on the underlying philosophy of the approach, and hence on the methods used to identify and evaluate structural similarity. Structural families derived, for example, using algorithms that search and cluster on the basis of common motifs will be different from those generated by procedures based on global structure comparison; and the results of such automatic procedures will differ again from those based on visual inspection, where software tools are used essentially to render the task of classification more manageable.
