2. Multiple sequence alignment in alignment programs such as ClustalX or Clustal Omega:
The sequences can either be pasted into the Web form or uploaded from a file to the Web form in the ClustalX program. Sequences can be aligned across their entire length (global alignment) or only in certain regions (local alignment). This is true for pairwise and multiple alignments. Global alignments need to use gaps (representing insertions or deletions) although local alignments can avoid them, by aligning regions between gaps.
The standard computational formulation of the pairwise problem is to identify the alignment that maximizes protein-sequence similarity, which is typically defined as the sum of substitution matrix scores for each aligned pair of residues, minus some penalties for gaps. This approach is generalized to the multiple sequence case by seeking an alignment that maximizes the sum of similarities for all pairs of sequences.
A substitution matrix describes the likelihood that two residue types would mutate to each other in evolutionary time. Understanding theories underlying a given matrix can aid in making proper choice. ClustalX can adopt four types of matrices :
- Point Accepted Mutation (PAM),
- Blocks Substitution Matrix (BLOSUM),
- GONNET, and
- DNA Identity Matrix (Unitary Matrix).
PAM matrices are traditionally amino acid scoring matrices, which refer to various degrees of sensitivity, depending on the evolutionary distance between sequence pairs. The BLOSUM matrices, also used for protein database search scoring (the default in Blastp), are divided into statistical significance degrees,
which are reminiscent of PAM distances. The BLOSUM matrices are most sensitive for local alignment of related sequences and are therefore ideal for identifying an unknown nucleotide sequence.
The GONNET matrix uses classical distance measures to estimate an alignment of the proteins. This data
is then employed to estimate a new distance matrix. This is useful to refine the alignment, estimate a new distance matrix, and so on. In the DNA Identity Matrix, you only get a positive score for a match, and a score of -10000 for a mismatch. As such a high penalty is given for a mismatch, no substitution is
allowed, although a gap may be permitted. A penalty is subtracted for each gap introduced into an alignment because the gap increases uncertainty into an alignment.
MultipleSequence Alignment using Clustal Omega:
https://www.ebi.ac.uk/Tools/msa/clustalo/
Upload or paste the sequence in the window:
Click Submit at the end of page. Leave all settings to default.
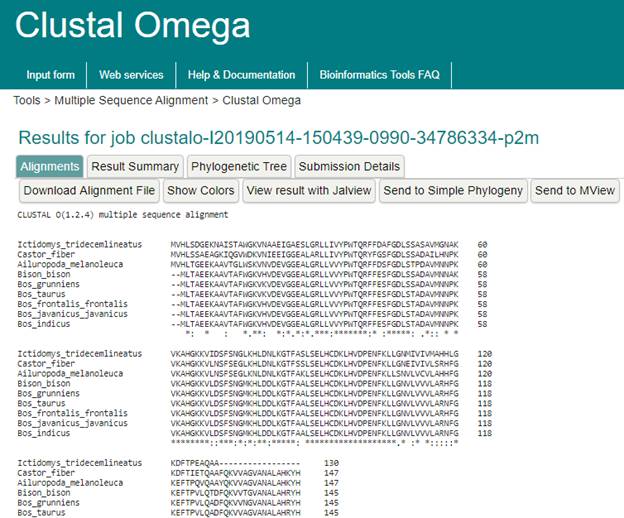
The Result
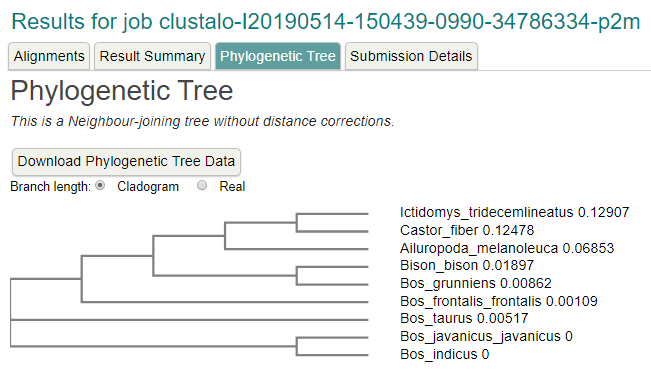
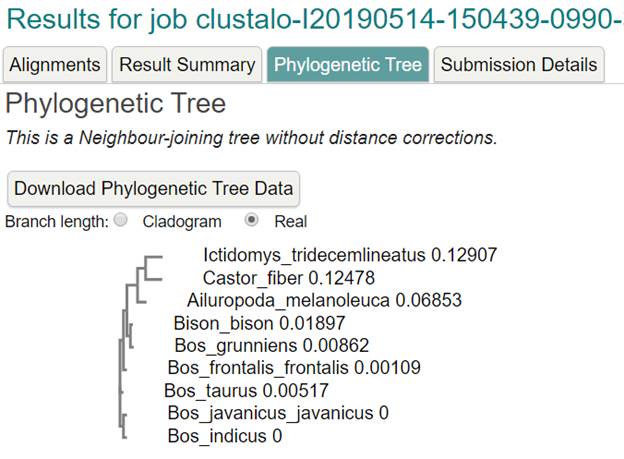
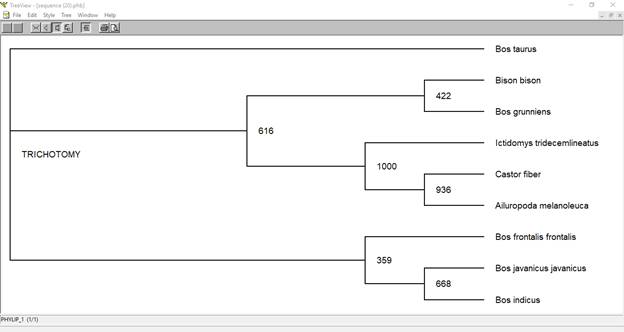
Tree using ClutalX and TreeView

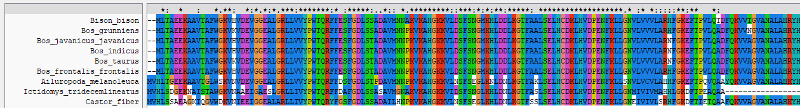
An * (asterisk) indicates positions which have a single, fully conserved residue.
A : (colon) indicates conservation between groups of strongly similar properties scoring > 0.5 in the Gonnet PAM 250 matrix.
A . (period) indicates conservation between groups of weakly similar properties scoring =< 0.5 in the Gonnet PAM 250 matrix.
Multiple Alignment using ClustalX2